It was penicillin that began losing battles almost as soon as medicine began winning them. Alexander Fleming pulled out a drug that could collapse pneumococcal and staphylococcal infections – the same medical triumph selected microbial survivors that no ward could easily restrain.
The paradox sits in plain view: antibiotics lengthened human life, and antibiotics trained bacterial populations to outlast the drugs meant to erase them. Penicillin rescued septic patients from ordinary injuries. But each exposure also rewarded any cell carrying a defensible mutation or borrowed resistance gene.
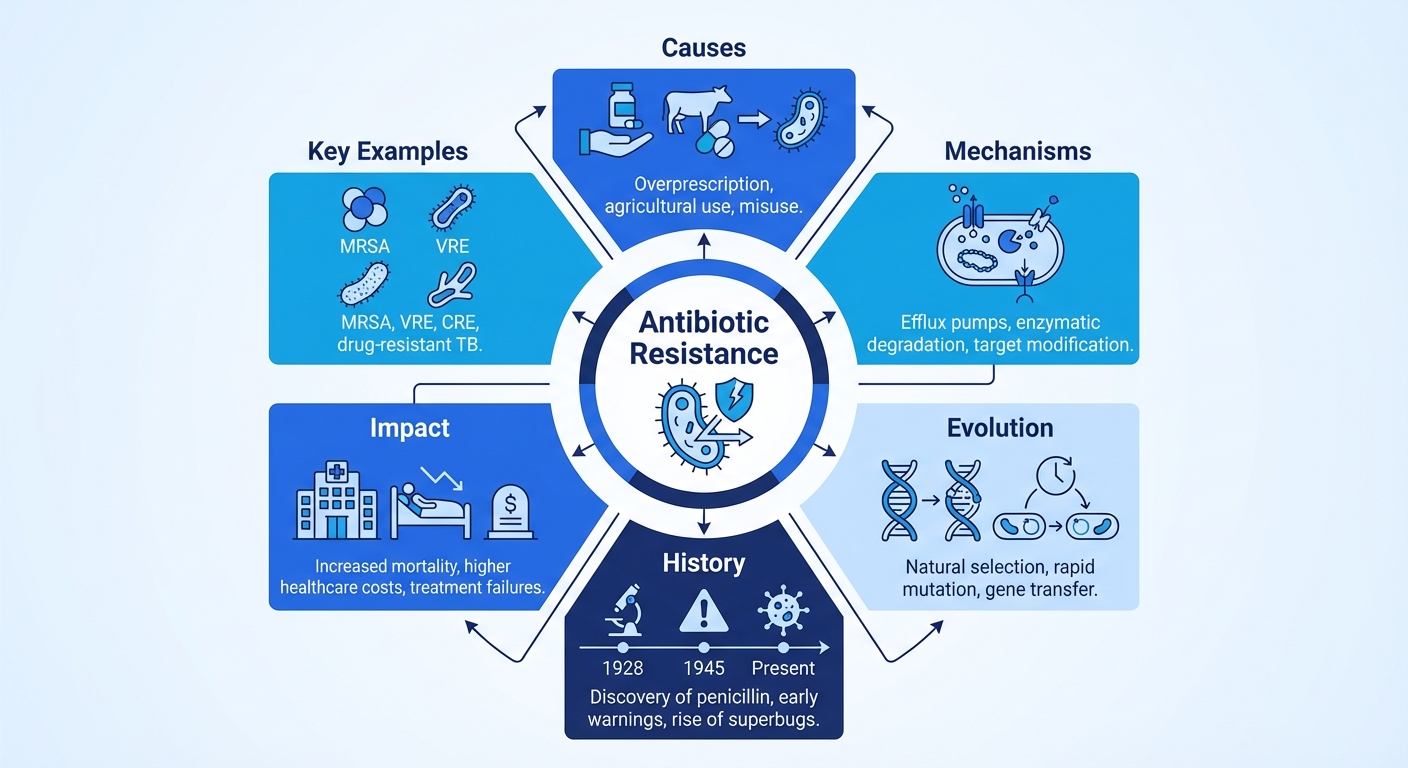
Rarely has a therapeutic breakthrough carried its liability so tightly fused to its benefit. Fleming warned in 1945 that low dosing would teach microbes, yet hospitals, pharmacies, farms, and ministries kept expanding use of antibiotics because the early cures looked too decisive to limit.
Penicillin didn’t create bacterial adaptation from nothing. Bacteria had been competing with organisms that make antibiotics for millions of years before any physician uncapped a vial. That old microbial arsenal later entered clinics under new pressure. Fast.
Carved out of wartime manufacturing, mass production of antibiotics turned a laboratory discovery into an entrenched protocol across civilian medicine, surgery, and military care, while bacterial selection pressure spread through every dose, every ward, and later every feedlot.
(American Chemical Society (acs.org), 1999 – acs.org/education/whatischemistry/landmarks/flemingpenicillin.html) In 2019, antimicrobial resistance was associated with nearly 5 million deaths worldwide, including at least 1.27 million deaths directly attributable to resistant infections. The World Health Organization now convenes repeated meetings on a problem that began at the same moment antibiotics became indispensable. The bargain still stands.
What is antibiotic resistance and what does it mean?
Entangled with every prescription, antibiotic resistance means a bacterium survives an antibiotic exposure that should have killed it or halted its replication. A drug still reaches the patient. The bacterium no longer yields.
Physicians see the consequence at the bedside when therapy that was standardized fails, infection persists, and the next treatment requires broader agents, longer admissions, toxic substitutions, or surgery that earlier decades often avoided. The same compounds that made care for pneumonia, trauma surgery, cancer chemotherapy, organ transplantation, and neonatal medicine feasible also created the selection pressure that rewards any microbe able to endure them.
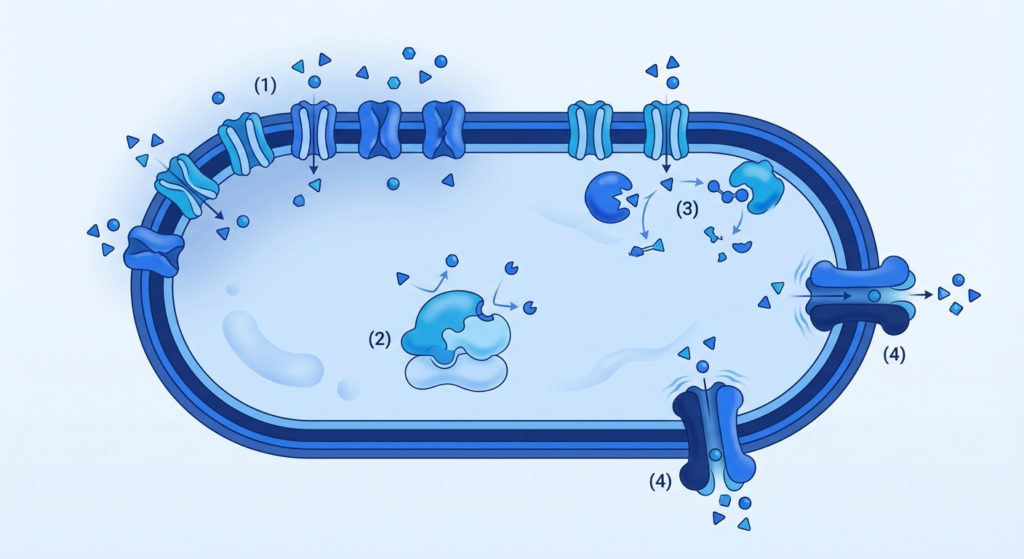
Forced by antibiotic contact, bacterial cells use several known mechanisms to preserve survival: they restrict drug access through altered membranes, they eject compounds through efflux pumps, they destroy drugs with enzymes, and they modify the molecular target that the antibiotic must bind.
(CDC (cdc.gov), 2025 – cdc.gov/antimicrobial-resistance/about/index.html) Staphylococcus aureus became a model for this pattern because clinicians watched it acquire resistance repeatedly across successive drugs, turning routine skin disease and bloodstream infection into recurrent therapeutic failure. Superbug remains a journalistic label, not a taxonomic class. More than 2.8 million antimicrobial-resistant infections occur each year in the United States – and those infections produce over 35,000 deaths.
The cure for one patient can strengthen the next patient’s adversary through selection among microbes.
Pressed by the old belief that resistance belonged only to hospitals, public health records later tied antibiotic resistance to community pathogens, use in agriculture, international travel, and other emerging infections that moved across borders faster than stewardship rules.
Antibiotic resistance doesn’t mean the human body rejected the drug; the bacterium altered the contest. That distinction matters. Each successful antibiotic course removes susceptible rivals and leaves the hardier minority with more ecological room, so the cure for one patient can strengthen the next patient’s adversary. The wording stays clinical. The biology doesn’t.
What are the 4 types of antibiotic resistance?
Spilled into four recurring categories, bacterial resistance to antibiotics operates through limiting drug uptake, modifying the drug target, inactivating the drug, and pumping the drug back out.
A Gram-negative cell can reduce antibiotic entry by altering porin channels in its outer membrane, so the compound reaches its intracellular target at sublethal concentration. A ribosome, a penicillin-binding protein, or a DNA gyrase can change shape enough to blunt binding of the antibiotic without stopping bacterial growth. Short answer: the bacterium blocks entry, alters the lock, breaks the key, or ejects the key.
- Limiting drug uptake – Bacteria alter membrane porins to reduce antibiotic entry, decreasing drug concentration at target sites.
- Modifying the drug target – Structural changes in ribosomes, penicillin-binding proteins, or DNA gyrases prevent effective antibiotic binding.
- Inactivating the drug – Enzymes such as beta-lactamases hydrolyze or modify antibiotics, rendering them ineffective before reaching their target.
- Pumping the drug out – Efflux pumps actively export antibiotics from the cell, lowering the intracellular drug concentration below therapeutic levels.
Dragged by direct molecular selection, each mechanism has named examples in clinical use. Beta-lactamases hydrolyze penicillins and cephalosporins before those drugs can bind cell-wall enzymes.
Tetracycline efflux pumps export drug molecules across the membrane and lower intracellular exposure. Vancomycin resistance in enterococci replaces the terminal D-Ala-D-Ala target with D-Ala-D-Lac – and that substitution sharply reduces binding affinity. Aminoglycoside-modifying enzymes add acetyl, adenyl, or phosphate groups to the drug and cripple antibacterial activity. One bacterium can carry more than one mechanism. Many do.
Anchored to success in treatment, these four tactics reveal the same old bargain in a harsher frame: every antibiotic that blocks a vital bacterial process also advertises a precise route for microbial escape, whether through exclusion, alteration, destruction, or export.
Clinicians often chase the phenotype with therapy that’s broader, therapy used in combination, or higher exposure, yet each escalation imposes fresh selection on the microbial population in the patient, on the ward, and far beyond the immediate infection site. The pharmacology saves lives. The microbiology keeps the invoice open.
When and how did antibiotic resistance first emerge?
Clutched at by early chemotherapy, antibiotic resistance emerged before penicillin became routine clinical treatment. It appeared mechanistically when bacteria neutralized drugs with enzymes or escaped killing through variation that was inherited.
Paul Ehrlich’s earlier antimicrobial work had already established the principle that microbes could stop responding to a once-lethal compound. During World War II, physicians observed sulfa drug resistance in treated infections, those failures exposed the same biological rule later attached to penicillin on a far larger scale. The warning arrived early.
Rarely has the standard origin story been so incomplete. Edward Abraham and Ernst Chain reported a penicillin-inactivating enzyme in 1940 – before penicillin entered widespread use – and that finding placed resistance beside the drug at birth rather than after its maturity.
In 1942, laboratories identified penicillin-resistant Staphylococcus aureus strains. This proved that bacterial populations could exploit the new pressure from therapy almost immediately after deployment in clinics began. Great Britain became one of the central settings for this recognition because wartime production, military demand, and hospital use created concentrated exposure in a compressed period.
Penicillin resistance was documented before penicillin was widely used in clinics.
Modern prescribing alone didn’t invent resistance. Studies of ancient permafrost and other ancient microbial reservoirs later detected resistance determinants that predated hospitals, patents, and fermentation on an industrial scale by millennia.
Use of antibiotics in humans amplified, sorted, and disseminated those capacities instead of creating them from nothing. That reframes the old bargain: the drug that saves the infected patient also serves as the sieve that enriches the hardiest surviving cell. The mechanism arrived first. The clinic accelerated it.
Penicillin resistance was detected in clinical bacteria within two years of penicillin’s widespread introduction, demonstrating that bacterial adaptation can occur almost immediately after new antibiotics enter use.
When did antibiotic resistance become a problem?
Antibiotic resistance became a recognized policy problem long before many governments treated it as an emergency.
Hospital wards had recorded difficult resistant infections for decades. But national and international authorities often handled them as technical microbiology issues rather than as systemic threats to routine medicine. Stuart Levy pushed that view into the open when he founded the Alliance for the Prudent Use of Antibiotics in the early 1980s, arguing that prescribing habits, agricultural exposure, and weak stewardship were already driving a wider crisis. The problem was old. The admission came late.
Rarely has institutional recognition lagged so far behind bedside evidence. The World Health Assembly adopted the WHO Global Action Plan on Antimicrobial Resistance on May 26, 2015 – decades after resistance had altered treatment pathways in hospitals and communities.
(World Health Assembly / WHO (who.int), 2015 – cdn.who.int/media/docs/default-source/antimicrobial-resistance/amr-spc-sel-glass/a68-r7-en.pdf?sfvrsn=fa7f3dde_2) Dame Sally Davies, serving as UK Chief Medical Officer, later described antibiotic resistance in language usually reserved for national catastrophe. That rhetoric reflected accumulated damage in the clinic rather than a fresh discovery. CDC surveillance then reported that six bacterial infections that were resistant to antimicrobials and started in hospitals increased by a combined 15% during the first year of the COVID-19 pandemic. Pressure exposed neglect.
Antibiotic resistance changed medicine for decades before the World Health Assembly called it an emergency.
Fractured into separate sectors, health systems treated antibiotics as immediate rescue tools and treated resistance as somebody else’s downstream problem – this kept the contradiction intact: every expansion of antimicrobial coverage protected one patient today while increasing selection pressure against the next patient’s options for treatment tomorrow.
Levy’s advocacy, the Alliance’s campaigns, and the World Health Assembly vote all acknowledged the same delayed truth that Fleming had already signaled in embryo. The threat became visible politically when institutions could no longer hide the clinical arithmetic. Still late.
Major milestones in the history of antibiotic resistance
Forced by each new drug launch, the history of antibiotic resistance moved through a brutal sequence of counter-moves rather than a steady march of therapeutic control. Not ideal.
Methicillin entered clinical practice in the early 1960s as a chemical workaround for penicillin resistance, and methicillin-resistant Staphylococcus aureus appeared in 1960, less than one year after introduction. The replacement failed early, and that event hardened a lesson that laboratories had already glimpsed with penicillin: each antibiotic class narrows one clinical danger while opening fresh evolutionary space for the next one.
- Penicillin introduction (1940s) – Transformed infection treatment, but resistance emerged almost immediately in hospital settings.
- Methicillin launch (1960) – Designed to overcome penicillin resistance, but MRSA appeared within one year, demonstrating rapid bacterial adaptation.
- Vancomycin reserve use (1960s – 1980s) – Held as a last-line agent for severe Gram-positive infections, but vancomycin-resistant Enterococcus surfaced by the mid-1980s.
- Superbug terminology (1966) – Entered public discourse as bacteria resistant to multiple antibiotics became a recognized clinical and societal threat.
Rarely has a milestone looked like progress for long. Tetracycline and chloramphenicol widened treatment options after the 1940s, macrolides and erythromycin followed in the 1950s, and hospitals treated each arrival as a renewed tactical advantage against bacterial disease.
By 1966, writers had already used the term superbug for bacteria resistant to multiple antibiotics – showing that resistance to multiple drugs entered public language far earlier than many modern accounts imply. Words changed. Bacteria moved faster.
MRSA appeared less than one year after methicillin, outpacing the drug meant to stop resistance.
Pushed into reserve status, vancomycin became a key therapy for serious Gram-positive infections in the 1960s, yet vancomycin-resistant Enterococcus emerged by the mid-1980s and showed that even drugs held back for severe cases could lose ground once ecological pressure built up inside hospitals.
Each milestone records the same reciprocal pattern: medicine modifies a molecule to restore control, and bacterial populations answer by selecting cells that bypass the modification. The chronology reads like innovation. The mechanism reads like pursuit without finish.
What is an example of an antibiotic resistant disease in history?
It was methicillin-resistant Staphylococcus aureus (MRSA) that became one of the clearest historical examples of an antibiotic-resistant disease.
MRSA turned ordinary staph infections into harder clinical problems because the organism resisted methicillin and often picked up resistance to other drugs used in hospital practice. Staphylococcus aureus had already established itself as a recurrent wound pathogen, bloodstream invader, and postoperative menace before methicillin resistance sharpened its significance. One organism kept returning.
Working in British hospital microbiology, Mary Barber identified penicillin-resistant Staphylococcus aureus in Great Britain by the late 1940s – her work documented how rapidly staphylococci adapted under antibiotic pressure in crowded clinical settings.
(British Medical Journal / PMC (bmj.com via ncbi.nlm.nih.gov), 1947 – pmc.ncbi.nlm.nih.gov/articles/PMC2056216) MRSA later extended that pattern. By 1974, fewer than 2% of staphylococcal infections in the United States were caused by MRSA, but by 2004 the figure had climbed to roughly 63%, showing how a marginal hospital strain could become a dominant therapeutic problem across a generation. But multidrug-resistant tuberculosis offers another historical case, MRSA remains the cleaner example because clinicians watched its ascent in direct sequence from one antibiotic era to the next.
Pressed by repeated substitutions for therapy, staph taught medicine the same lesson at multiple intervals: a new drug can suppress yesterday’s resistant strain and still prepare the ground for tomorrow’s harder descendant.
MRSA didn’t merely resist one molecule; it exposed the liability built into rescue antibiotics used at scale, because every successful course against susceptible staphylococci enriched the ecological position of the variants left standing. Surgeons, intensivists, and ward teams then paid the price through longer admissions, narrower margins for therapy used in empirical cases, and recurrent outbreaks. The patient changed. The pattern didn’t.
How resistant bacteria have evolved since the discovery of antibiotics
Dragged by antibiotic pressure, resistant bacteria have evolved through mutation, gene exchange, clonal expansion, and ecological opportunism rather than through a slow, isolated drift inside single species.
Penicillin entered medicine after its 1928 discovery, yet penicillin-resistant Staphylococcus aureus was documented by 1944 – just sixteen years after Fleming’s first observation – showing how quickly selection could favor existing survival traits once treatment became common. Bacteria didn’t wait. They adapted fast, as each antimicrobial victory removed susceptible competitors and widened the niche for survivors carrying a defense.
- Mutation – Spontaneous genetic changes confer resistance, especially under antibiotic selection pressure.
- Horizontal gene transfer – Plasmids and transposons allow bacteria to share resistance genes across species and genera.
- Clonal expansion – Resistant strains proliferate rapidly when antibiotics kill off susceptible populations.
- Ecological opportunism – Bacteria exploit new environments created by medical, agricultural, or environmental antibiotic use.
Rarely has bacterial evolution depended on inheritance alone. Plasmid-mediated horizontal transfer of resistance was first demonstrated in Japan in the early 1960s, and that finding showed that one bacterium could pass resistance determinants sideways to another rather than waiting for line-by-line descent through generations.
Plasmids enabled sudden jumps across strains and sometimes across species. Hospitals, farms, sewage systems, and international transport routes became exchange chambers for genes for resistance. Neisseria gonorrhoeae then offered a brutal textbook case, evolving resistance after each major antibiotic directed at it – from sulfonamides in the 1930s through later agents introduced to regain clinical control.
Bacteria shared resistance genes sideways, not just by inheritance, accelerating adaptation across species.
It was vancomycin that many clinicians treated as a durable backstop after its 1958 clinical introduction, yet clinically important vancomycin-resistant enterococci emerged in the late 1980s and dismantled that assumption inside high-antibiotic hospital environments.
Methicillin followed the same script after its 1960 arrival, as MRSA spread from a targeted workaround into a pathogen defined by hospitals. The pattern stayed constant: medicine changed the molecule, bacterial populations changed the terms of survival. Evolution kept billing the cure.
Are humans becoming more resistant to antibiotics?
It is bacteria and fungi, not humans, that become resistant to antibiotics. A clinician gives the drug to a person, but the microbe changes its biology and survives exposure that once killed it or stopped replication.
The patient doesn’t acquire antibiotic resistance the way a pathogen does. The patient instead carries a resistant infection, fails first-line treatment, or serves as a host through which resistant organisms spread to other people. That distinction matters.
Embedded in CDC language, the mechanism stays blunt: antimicrobial resistance happens when germs such as bacteria and fungi defeat the drugs designed to kill them.
(CDC (cdc.gov), 2025 and WHO (who.int), 2025 – cdc.gov/antimicrobial-resistance/about/index.html) The World Health Organization states the practical outcome with equal bluntness – when antimicrobial resistance develops, antibiotics lose their punch and infections become difficult or impossible to treat. Travel and trade then move resistant organisms across borders, so a strain selected in one hospital, farm system, or city can appear in Europe or elsewhere through ordinary human movement. More than 2.8 million antimicrobial-resistant infections occur in the United States each year. The host is human. The adaptation is microbial.
Carved into clinical misunderstanding sits a recurring error: patients often say their bodies “got used to” antibiotics, and some members of the medical profession still tolerate that shorthand, but the phrase hides the central hazard. It relocates the biological change from the organism causing disease to the person receiving care.
That error matters deeply. If physicians and patients misplace the adaptation, they also misplace responsibility for stewardship, surveillance, infection control, and preservation of drugs. The same medicine that protects a human host from sepsis, pneumonia, or postoperative collapse still selects for the microbe that can survive the next course. The body survives. The germ learns.