Embedded in the mortality ledger of the pre-antibiotic world, bacterial infection erased millions who would now survive on a short ward course of penicillin or one of its descendants. In industrialized countries before antibiotics, average life expectancy at birth sat near 47 years. By the end of the 20th century in the United States, it reached 78.8 years – with antibiotics carrying part of that gain. Penicillin opened that ascent, yet the same discovery that turned lethal sepsis into a treatable event also gave bacteria the selective pressure that would train resistance against every successor.
Cure and adaptation came from the same encounter. What Fleming exposed in a contaminated culture dish – a moldy petri plate left too long on his lab bench – didn’t merely clear infection, it also unlatched a permanent contest between microbial killing and microbial escape.
The discovery of penicillin and Alexander Fleming’s breakthrough
Spilled into a neglected culture plate, Penicillium notatum gave Alexander Fleming the observation that medicine had failed to manufacture by intention in 1928. Fleming saw a mold contaminate his staphylococcal plate and leave a clear inhibition zone where bacterial growth collapsed. He identified in that accident a substance later named penicillin. The image became canonical.
But the plate didn’t cure patients. Fleming published the finding, yet his laboratory couldn’t purify penicillin into stable, measurable doses for use in treatment – a failure that delayed broad application while preserving the biological contest already latent in the mold’s chemistry. The same molecule that opened bacterial killing also prepared the ground on which bacteria would later harden evasive tactics.
In Fleming’s hands, penicillin remained more laboratory curiosity than ward instrument, because extraction, concentration, and storage imposed chemical obstacles that his bench could not overcome with available methods. Howard Florey, Ernst Boris Chain, and Norman Heatley at Oxford later built the procedures that converted the observation into a usable drug. They scaled culture. Refined crude filtrates. Assembled enough material for application in clinics under wartime strain.
Fleming didn’t develop penicillin for treatment – Oxford did. In 1945, the Nobel Prize in Physiology or Medicine went jointly to Fleming, Florey, and Chain, an award that acknowledged both the solitary plate and the institutional machinery required to make the plate matter. Penicillin’s breakthrough therefore arrived as a split event: one man recognized the inhibition zone, then another team rendered the mold’s lethal gift deployable for use in medicine, after which microbial adaptation acquired an adversary strong enough to select against.
Fleming’s discovery alone didn’t cure anyone; penicillin became medicine only after Oxford industrialized it.
Under wartime pressure, hospitals received penicillin as a near-miraculous anti-infective. The miracle depended on fermentation vats, protocols used in hospitals, animal testing, and rationed distribution rather than on serendipity alone.
Patients with septic wounds, puerperal infections, gonorrhea, and pneumococcal disease now faced organisms with a new biochemical obstacle in their path, because penicillin blocks the synthesis of bacterial cell walls by binding penicillin-binding proteins and disrupting peptidoglycan cross-linking. Bacteria answered later. The delay deceived clinicians. Penicillin didn’t end the contest with microbes – penicillin made it more intense by making bacterial survival contingent on resistance mechanisms, a pressure that began the moment clinical success became repeatable.
Penicillin’s introduction transformed hospital mortality, but also began the evolutionary arms race that defines modern therapy for infectious disease.
What was the first antibiotic in history?
It was Salvarsan that entered medicine in 1909 as the first antimicrobial drug, years before Alexander Fleming discovered penicillin in 1928. Paul Ehrlich developed Salvarsan as a treatment for syphilis, and physicians used it as a targeted chemical assault against Treponema pallidum rather than as a broad antibacterial agent. The chronology unsettles the common shorthand that names penicillin first.
Penicillin was the first widely used natural antibiotic. Not the first antimicrobial intervention to reach practice in clinics. That distinction matters.
Across German industrial chemistry, Bayer developed Prontosil in 1932 or 1933, and Gerhard Domagk established its antibacterial value after Josef Klarer contributed to its synthesis. (Source: Science History Institute, 2026 – sciencehistory.org/education/scientific-biographies/gerhard-domagk) Prontosil became the first commercially available sulfa drug, placing a synthetic compound – not a mold derivative – at the center of early mass antibacterial therapy.
Hospitals adopted sulfonamides before penicillin reached broad wartime distribution because chemists could manufacture them more reliably than early penicillin preparations. Industry drove that phase. Ehrlich’s Salvarsan therefore served as a precursor, and Domagk’s Prontosil proved that antimicrobial chemotherapy could scale before fungal metabolites became clinically dominant.
By naming penicillin as the beginning, public memory compresses a fractured lineage into one emblem and hides the older fact that chemistry on a factory scale reached patients before mold broth did. That compression also masks the central liability threaded through the field from the start: every stronger antimicrobial method increases the selective burden on microbes and narrows future room for treatment.
Salvarsan initiated directed killing. Prontosil made it chemistry on a factory scale. Penicillin later amplified it with greater force and wider reach, and each success strengthened medicine while teaching pathogens that survival would depend on adaptation under chemical siege.
Treating infections before antibiotics existed
In operating theaters before penicillin, surgeons fought bacteria with steel, drainage, cautery, antiseptic washing, and amputation rather than with a pill or vial. Joseph Lister introduced antiseptic surgery in the late 1860s, and surgeons used carbolic acid and disciplined wound handling to prevent contamination from gaining an irreversible hold in tissue.
Patients survived some wounds because knives removed pus pockets and dead matter before invasion of the bloodstream accelerated. Many didn’t. Doctors could sometimes control the source of infection, yet they could rarely extinguish organisms once dissemination began – a limitation that made the later antibiotic era look like liberation while embedding the pressure that would later educate microbes.
By 1890, Emil von Behring and Shibasaburo Kitasato had discovered antitoxin serum therapy for diphtheria, and by September 1894 Emile Roux reported clinical results that spread its use across hospitals. Serum therapy didn’t kill bacteria directly; serum therapy neutralized toxins that bacteria produced, which meant physicians could reduce mortality in selected diseases without possessing a true antibacterial drug.
Ancient Sudanese Nubia complicates the clean chronology. Human skeletal remains from 350 – 550 CE contain traces of tetracycline – indicating that human populations encountered antibiotic compounds long before laboratories named them – isolated them, or prescribed them under unswerving measurements. Accidental exposure came first. Systematic control arrived later, and the same biological antagonism that once leaked from fermented material would later be weaponized on a scale that bacteria couldn’t ignore.
Ancient bones show exposure to tetracycline, centuries before anyone purified or prescribed antibiotics.
At the bedside, doctors relied on entrenched protocols that mixed isolation, wound debridement, topical antiseptics, serum where available, and physical removal of infected tissue because common bacterial infections still killed with routine efficiency.
Pneumonia killed. Tuberculosis persisted. Gonorrhea maimed. Even when an intervention worked, it often worked by limiting spread rather than by sterilizing the patient, and that distinction governed medicine before antibiotics: physicians constrained damage more often than they eliminated cause. The arrival of antibiotics didn’t invent microbial antagonism – it industrialized an old biological fact and turned selective pressure into standard care.
What happened before antibiotics were invented?
Before antibiotics, physicians treated infection with surgery, antiseptics, isolation, and supportive care. Common bacterial diseases often remained lethal. Pneumonia, tuberculosis, gonorrhea, puerperal sepsis, wound infections, and meningitis imposed mortality that no clinician could reliably reverse once organisms moved beyond a localized focus.
Doctors opened abscesses, drained empyemas, excised gangrenous tissue, and amputated infected limbs because mechanical removal of the burden of bacteria offered the only immediate chance of arresting spread. Those methods were brutal. Those methods were rational. Before penicillin entered practice after Fleming’s 1928 discovery at St. Mary’s Hospital in London, medicine managed infection largely by containment and tissue sacrifice rather than by direct biochemical attack on bacteria.
Across the years before penicillin, sulfonamides provided the first widely effective antibacterial drugs, with Prontosil reaching use in the 1930s before penicillin achieved broad availability. That sequence unsettles the common simplification that places one clean break in 1928 and ignores the transitional decade in which synthetic drugs reduced deaths from infections caused by streptococcal and other bacteria.
At Dakhleh Oasis in Egypt, archaeological evidence has also suggested ancient tetracycline exposure in late Roman-period remains – paralleling the better-known findings from Sudanese Nubia – indicating that antimicrobial compounds touched human bodies long before laboratories produced standardized therapy. Ancient exposure didn’t create medicine. But ancient exposure did show that bacterial antagonists existed in human environments long before clinicians learned to measure, purify, and deploy them with consistency.
After antibiotics arrived, hospitals could finally strike the pathogen rather than carving away the consequences, yet the older era had already exposed the durable structure of the conflict.
Every pre-antibiotic intervention that removed infected material without eradicating microbial life revealed how difficult control of bacteria would remain even after chemistry improved. Sulfonamides and penicillin changed the odds, not the underlying contest. The benefit was immense, the liability came attached: once medicine could attack bacteria directly and at scale, bacteria faced a recurrent filter that rewarded resistant survivors. This converted a triumph in use for therapy into an enduring evolutionary arms race that pre-antibiotic physicians never had the power to trigger so intensely.
Before antibiotics, even minor infections often resulted in death or permanent disability, as medicine lacked any effective means to kill bacteria within the body.
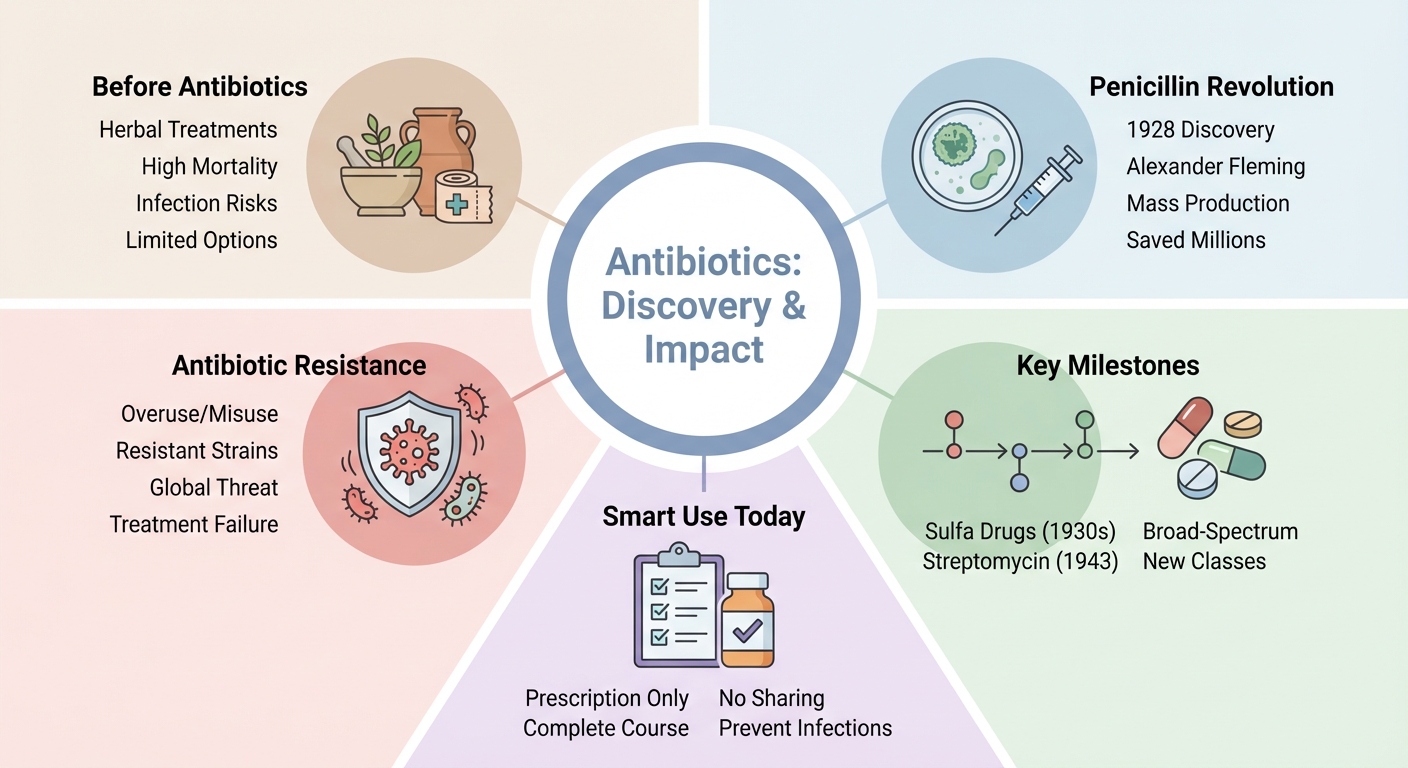
The golden age of antibiotic discovery
It was soil microbiology that supplied the antibiotic boom more than any single genius after Fleming’s mold plate had altered clinical ambition. The golden era of antibiotic discovery ran from the 1950s to the 1970s, when laboratories and pharmaceutical firms screened actinomycetes and other microorganisms for compounds that could suppress, stall, or kill pathogenic bacteria across multiple infection sites.
Streptomyces species became assets for chemistry on a factory scale. Petri dishes turned into extraction programs, the field no longer waited for accident but hunted antagonism in dirt, fermentation broth, and microbial competition – each newly isolated weapon widened treatment while tightening the evolutionary vise on bacterial survivors.
In 1945, Benjamin Minge Duggar discovered chlortetracycline, marketed as Aureomycin, the first tetracycline antibiotic, from a Streptomyces-derived source that expanded the practical logic of broad-spectrum therapy. Streptomycin, isolated earlier from Streptomyces griseus, had already shown that soil bacteria could furnish drugs against severe infections such as tuberculosis (Source: Journal of Biological Chemistry, 1944), shifting discovery from chance contamination toward organized screening platforms.
Penicillin didn’t dominate this era alone. Actinomycetes did. Companies built pipelines around culture collections, bioassays, fermentation engineering, purification chemistry, and scalable manufacturing because natural-product prospecting yielded repeated clinical prizes under discipline in industry. Success bred haste. And haste often treated each new compound as inexhaustible before bacterial populations had even begun to register the selection pressure imposed by mass use.
Soil bacteria, not only molds, became the main source of new antibiotics after penicillin.
Across postwar hospitals, antibiotic abundance changed physician behavior as much as it changed microbial mortality, because drugs that once looked like scarce salvage became routine first-line prescribing tools for febrile patients, postoperative wounds, and respiratory infections. These drugs also saw use for prophylaxis around procedures.
The abundance concealed a structural limit – screening recovered many molecules from related ecological niches, and organisms in those niches had long exchanged chemistry on a factory scale with countermeasures. This meant resistance wasn’t an aberration arriving after victory but a delayed invoice attached to victory from the beginning. The golden age generated breadth for therapy, yet each expansion of the armamentarium also increased the probability that medicine would consume its own leverage faster than institutions could replace it.
Key scientists and major milestones in antibiotics history
Forced by the flood of new compounds after the golden era began, scientific credit fractured across discoverers, chemists, industrial engineers, and institutions that made one another indispensable. Selman Waksman coined the term “antibiotics” in 1942, giving the field its durable label before its boundaries had fully stabilized in laboratory or clinic.
Albert Schatz discovered streptomycin in Waksman’s laboratory in 1943, and that drug became the first antibiotic effective against tuberculosis – a disease that had killed roughly 1 in 7 people who contracted it. Names mattered. Molecules mattered more. Each milestone expanded reach for therapy, yet each new class multiplied the number of microbial encounters in which survival could reward resistance rather than eradication.
In 1945, Dorothy Crowfoot Hodgkin solved the structure of penicillin by X-ray crystallography, converting a clinically potent but chemically elusive substance into a defined molecular object that chemists could manipulate with far greater precision.
This structural knowledge altered manufacturing, storage, derivative design, and quality control because laboratories could now align purification and synthesis with a resolved scaffold instead of an empirical extract alone. Giuseppe Brotzu first isolated the parent cephalosporin compound in 1948 from material linked to Sardinia, and that work opened another lineage of beta-lactam development beyond penicillin. But Nobel memory omitted one central discoverer. Albert Schatz did not share Waksman’s 1952 Nobel Prize, despite Schatz’s direct role in the discovery of streptomycin. Credit followed institutions unevenly.
Albert Schatz discovered streptomycin but wasn’t given the Nobel Prize awarded to Waksman.
At Peoria, Andrew J. Moyer and the USDA Northern Regional Research Laboratory improved large-scale penicillin production methods – work that started with corn steep liquor as a cheap nutrient source – showing that milestone history belongs as much to process engineering as to bench revelation.
A vial requires systems. A Nobel medal doesn’t. The field advanced through renamed concepts, isolated organisms, crystallographic resolution, refinement through fermentation, and compounds that were clinically decisive and made bacterial disease less sovereign over human survival. The same chain of milestones also tightened medicine’s dependence on finding new drugs, because every successful antibiotic shortened its own uncontested lifespan by exerting selective force on the organisms it was built to kill.
- Selman Waksman – coined the term “antibiotics” in 1942, establishing the conceptual foundation for the field
- Albert Schatz – discovered streptomycin, the first drug effective against tuberculosis, in 1943
- Dorothy Crowfoot Hodgkin – solved the structure of penicillin in 1945, enabling chemical refinement and mass production
- Giuseppe Brotzu – isolated the parent cephalosporin compound in 1948, opening a new beta-lactam lineage
- Andrew J. Moyer & USDA NRRL – developed industrial-scale penicillin production, turning laboratory success into clinical reality
When did people start taking antibiotics?
People started taking antibacterial drugs in the 1930s with sulfonamides, and they started taking penicillin widely during World War II after mass production succeeded. Sulfonamides arrived first as the earliest broadly used antibacterial medicines available to ordinary patients beyond isolated experimental settings.
Penicillin then moved from scarce laboratory product to routine wartime agent for therapy when deep-tank fermentation and scaling on an industrial level delivered enough material for military and civilian medicine. Timing mattered. Access mattered more. A discovery in a paper doesn’t create a treatment era until factories can fill vials at volume, physicians can prescribe them beyond exceptional cases.
During World War II, deep-tank fermentation enabled mass production of penicillin – using 10,000-gallon tanks that dwarfed earlier laboratory flasks. Wartime demand accelerated cooperation between laboratories, manufacturers, and government-linked production programs.
Pharmaceutical firms converted microbial growth into output for industry by controlling aeration, nutrient supply, contamination risk, and downstream extraction, all of which had previously prevented penicillin from reaching patients in adequate quantity. Soldiers with wound infections, pneumonia, septicemia, and gonorrhea received penicillin on a scale impossible in the 1930s. Factories changed medicine. Before that expansion, sulfonamides had already represented the first major practical step into routine antibacterial treatment, proving that patients began taking antibacterial drugs before penicillin became the iconic agent of the category.
Once antibiotics entered ordinary prescribing, bacterial infection ceased to be a fixed sentence in many cases and became a problem that clinicians expected to reverse.
That expectation restructured hospitals, surgery, obstetrics, and military medicine, but it also normalized repeated exposure of microbial populations to drugs that could no longer remain ecologically invisible. The benefit arrived in bodies saved, amputations avoided, and pneumonias survived. The liability entered with the same prescription pad, because widespread intake converted therapy using antimicrobials from rare intervention into a constant evolutionary pressure, and bacteria learned under that pressure every time human systems succeeded in delivering treatment at scale.
How different classes of antibiotics developed over time
Entangled with the naming of disease victories, antibiotic classes emerged as a sequence of chemical solutions to distinct bacterial vulnerabilities rather than as one uninterrupted penicillin story. Sulfonamides formed the first widely used antibacterial drug class after Gerhard Domagk discovered Prontosil in 1932 – the first synthetic compound that worked against bacterial infections – and clinicians used those synthetic agents before penicillin became broadly available.
Penicillins then launched the beta-lactam era after Alexander Fleming’s 1928 discovery, with benzylpenicillin moving into clinical use and later derivatives such as amoxicillin extending oral practicality and spectrum. Chronology mattered. Mechanism mattered more. Each class targeted bacteria through a different biochemical route, and each route widened treatment while creating another selective corridor through which resistant organisms could pass.
- Sulfonamides – first widely used synthetic antibacterial drugs, targeting folate synthesis in bacteria
- Penicillins – natural beta-lactam antibiotics, disrupting bacterial cell wall synthesis
- Aminoglycosides – such as streptomycin, inhibiting bacterial protein synthesis by binding the 30S ribosomal subunit
- Tetracyclines – broad-spectrum antibiotics, blocking tRNA binding to the bacterial ribosome
- Macrolides – interfering with protein synthesis via the 50S ribosomal subunit, effective against respiratory pathogens
- Cephalosporins – beta-lactam antibiotics with broader spectrum and greater beta-lactamase stability than penicillins
- Carbapenems – advanced beta-lactams, highly resistant to most beta-lactamases, reserved for severe infections
- Quinolones/Fluoroquinolones – synthetic agents targeting bacterial DNA gyrase and topoisomerase IV
- Oxazolidinones – such as linezolid, inhibiting protein synthesis initiation in resistant Gram-positive bacteria
By the 1940s, new natural-product classes expanded rapidly: streptomycin entered as the first aminoglycoside, tetracyclines followed, and macrolides soon joined the antibacterial armamentarium with distinct ribosomal targets. These drugs reached across respiratory disease, tuberculosis, enteric infections, and mixed community pathogens.
Cephalosporin C was discovered in 1948, yet the first cephalosporin drugs reached practice later, which undercuts the easy assumption that discovery and use in routine practice move together on one timetable. Molecules wait. Manufacturing, toxicity screening, pharmacokinetics, formulation, and clinical demand determine when a class becomes real at the bedside – not just when someone pulls it from a culture plate. Cephalosporins later became a major beta-lactam family, and carbapenems pushed that family toward greater stability against many beta-lactamases, though never toward permanent security.
Cephalosporins were discovered in 1948, but use in medicine lagged behind by years.
Under pressure from resistant organisms and therapeutic gaps, later decades produced wholly synthetic or heavily modified agents such as ciprofloxacin and linezolid. These drugs showed that antibiotic history did not remain confined to soil extracts and fungal metabolites. (Source: PubMed / Pharmacotherapy, 2003 – pubmed.ncbi.nlm.nih.gov/12847568)
Chemists redesigned scaffolds, altered side chains, and pursued intracellular penetration, oral bioavailability, serum half-life, and Gram-negative activity with increasing precision because older classes were losing ground in hospitals and clinics. The pattern kept repeating. A new class solved yesterday’s failure, then usage selected tomorrow’s workaround in bacterial populations, leaving finding new drugs less like a ladder of final answers than a rolling technical reprieve whose duration kept shortening.
Antibiotic resistance and the discovery pipeline crisis
Carved out by decades of successful prescribing, antibiotic resistance now confronts medicine as the predictable biological response to antibiotic abundance rather than as an anomalous postscript. (Source: PMC / Clinical Infectious Diseases review, 2019 – pmc.ncbi.nlm.nih.gov/articles/PMC6749829) By 1944, gonorrhea treatment failures with sulfonamides exceeded 30 percent – this means large-scale clinical resistance appeared early, before the antibiotic age had even matured into its self-congratulatory mythology.
The warning was visible. Institutions kept prescribing anyway. Hospitals, farms, clinics, and pharmaceutical markets treated antibacterial efficacy as a renewable resource, although every dose imposed selection on bacteria and favored survivors carrying beta-lactamase production, target modification, efflux capacity, permeability changes, or transferable genes for drug resistance.
In 1976, plasmid-mediated penicillin resistance in Neisseria gonorrhoeae was reported, marking a turning point because bacteria had not merely mutated under pressure but had also acquired mobile genetic machinery that could spread resistance between strains with ruthless efficiency. (Source: PubMed / Antimicrobial Agents and Chemotherapy, 1977 – pubmed.ncbi.nlm.nih.gov/404964)
Methicillin-resistant Staphylococcus aureus, vancomycin-resistant enterococci, and carbapenem-resistant Enterobacterales now anchor hospital concern because each organism undermines drugs once reserved for severe or last-line use. The newest crisis isn’t resistance alone – the pipeline is thin, discovery programs slowed as scientific difficulty rose, and stewardship tightened sales volume. Generic competition compressed profit, and companies retreated from antibacterial development despite the escalating clinical need that their withdrawal made more dangerous.
By 1944, sulfonamide resistance in gonorrhea exceeded 30 percent, before use of penicillin in a broad sense.
- Beta-lactamase production – enzymes that break down penicillins and cephalosporins, rendering them ineffective
- Target site modification – changes in bacterial proteins that antibiotics bind, reducing drug efficacy
- Efflux pumps – bacterial systems that expel antibiotics before they can act
- Plasmid-mediated resistance – genetic elements that transfer resistance between bacteria, accelerating spread
Under federal surveillance, the CDC’s 2019 Antibiotic Resistance Threats Report presented national estimates for 18 antimicrobial-resistant bacteria and fungi, evidence of a threat large enough to need centralized counting rather than anecdotal alarm.
The list itself indicts the system. A mature field of use for therapy shouldn’t need recurrent salvage against organisms that older drugs once controlled. So antibiotic medicine occupies a trap of its own construction: the more widely it succeeds, the faster it consumes the conditions that made that success possible, and the fewer commercial actors remain willing to replenish the armamentarium.
| Entity | Resistance problem | Clinical setting | Historical significance |
|---|---|---|---|
| Methicillin-resistant Staphylococcus aureus (MRSA) | Resistance to many beta-lactam antibiotics | Hospitals and community settings | Demonstrated that resistance could neutralize standard anti-staphylococcal therapy |
| Vancomycin-resistant enterococci (VRE) | Resistance to vancomycin | Hospitals, especially high-acuity wards | Compromised a drug used when other agents had failed |
| Carbapenem-resistant Enterobacterales (CRE) | Resistance to carbapenems, often last-line agents | Hospitals and long-term care facilities | Exposed the fragility of the late-stage antibiotic reserve |
| Neisseria gonorrhoeae | Early sulfonamide failure; later plasmid-mediated penicillin resistance | Sexually transmitted infection treatment programs | Provided one of the earliest large-scale demonstrations that resistance could outpace routine therapy |
Natural origins versus synthetic development in antibiotic history
Under the famous mold story, antibiotic history never belonged to nature alone or to chemistry alone. Both lineages built the modern anti-infective armamentarium by different routes toward the same bacterial target. Penicillin came from Penicillium and established the power of natural products, while antimicrobial chemotherapy had already advanced through synthetic compounds such as Salvarsan and later sulfonamides under Paul Ehrlich’s “magic bullet” logic of selective toxicity.
Laboratories didn’t choose one lineage and discard the other. They combined them, modified them, screened them, and industrialized them. Natural discovery supplied many scaffolds, synthetic development supplied tunability and manufacturability – the distinction remained porous as semisynthetic drugs joined both traditions inside one economy for therapy.
A toxicity disaster hardened federal drug regulation around one of the earliest synthetic antibacterial agents, not a therapeutic success. (Source: FDA, 2026 – fda.gov/about-fda/histories-product-regulation/sulfanilamide-disaster) The 1937 Elixir Sulfanilamide disaster killed more than 100 people in the United States after a sulfanilamide preparation used diethylene glycol as solvent. The deaths exposed lethal weaknesses in safety control before the product hit the market.
Congress enacted the Federal Food, Drug, and Cosmetic Act in 1938 in response, requiring stronger evidence of drug safety before marketing and enlarging the Food and Drug Administration’s regulatory authority. Chemistry on a factory scale offered scale. But synthetic chemistry also exposed the cost of inadequate oversight. Nature didn’t exempt drugs from danger either, but the industrial manipulation of antimicrobial agents forced governments to confront the fact that life-saving compounds and mass poisoning could emerge from adjacent benches.
A drug meant to save lives killed over 100 people, forcing stricter regulation on antibiotics.
- Natural products – penicillins, cephalosporins, tetracyclines, and macrolides, derived from molds or soil bacteria, provided initial scaffolds
- Synthetic agents – sulfonamides, quinolones, and oxazolidinones, designed or modified by chemists for improved efficacy and manufacturability
- Semisynthetic derivatives – modified natural antibiotics such as amoxicillin or ceftriaxone, combining biological origin with chemical tuning
- Regulatory milestones – safety disasters like Elixir Sulfanilamide led to new drug approval laws, shaping future antibiotic development
Across the long arc from Ehrlich’s selective chemicals to fungal metabolites, cephalosporins, tetracyclines, quinolones, and oxazolidinones, the field kept oscillating between finding antibacterial power in organisms and building it through design.
Screening programs searched soil, broth, and fermentation residues for useful antagonists; medicinal chemists then altered side chains, improved absorption by mouth, reduced toxicity, and attempted to outrun resistance with derivative after derivative. The contradiction never closed. The same ingenuity that extracts cures from mold or makes them from concept also widens their use – intensifying the selective burden on bacteria, and leaving medicine in the same old bind identified at the beginning: every advance against infection carries within it the mechanism that erodes the advance.